GEN – Genetic Engineering & Biotechnology News
Dual-Vector System Generates CAR T Cells In Vivo

Manufacturing CAR T cell therapy—one of the most powerful weapons against certain blood cancers—has to date involved an elaborate process, through which doctors first extract a patient’s immune cells and ship them to a specialized facility where the cells are genetically reprogrammed to fight cancer. The engineered cells must then be shipped back, so they can be infused into the patient’s bloodstream. The process can take weeks and may cost hundreds of thousands of dollars, placing it out of reach for many of the patients who need it most.
Scientists at UC San Francisco and collaborators have now reported on the development of a method to precisely reprogram these cancer-fighting cells directly inside the body, potentially eliminating the manufacturing process, cost, and waiting time.
![Justin Eyquem, Associate Professor of Medicine at UCSF. [Susan Merrill/UCSF]](https://www.genengnews.com/wp-content/uploads/2026/03/UCSF_20240524_Pew-Award-Eyquem_0028-300x192.jpeg)
It is the first time that scientists have integrated a large sequence of DNA at a specific site in human T cells without removing the cells from the body. “To our knowledge, this is the first demonstration of programmable, site-specific integration of a large DNA payload into T cells in vivo,” Justin Eyquem, PhD, an associate professor of medicine at UCSF, told GEN. “While others have pursued in vivo CAR T cell generation using either lipid nanoparticles for transient mRNA delivery or engineered lentiviral vectors for random integration, we achieved a combination of cell specificity, locus specificity, and stable integration of a large transgene in a living organism.”
Crucially, in reported tests the new, targeted approach was found to outperform the standard method of randomly integrating DNA using viruses. And in experiments using mice with humanized immune systems, the researchers used the new technology to successfully treat aggressive leukemia, multiple myeloma, and even a solid tumor. “… the most exciting, and honestly the most surprising, finding was how well the approach worked in solid tumors,” Pierre-Louis Bernard, PhD, a postdoc in the Eyquem lab, acknowledged to GEN.
“There has been a broadly held assumption in the field, shared by our reviewers, that engineering a small number of T cells circulating in the blood and expecting them to traffic to a solid tumor, encounter antigen only upon arrival, and then expand sufficiently to control the tumor would be an extremely tall order. We proved it was possible. That result, more than any other, convinced us that the platform has therapeutic reach well beyond hematological malignancies.”
Bernard, is co-first author, and Eyquem senior and corresponding author of the team’s published paper in Nature titled, “In vivo site-specific engineering to reprogram T cells.”
CAR T cell therapy works by giving T cells—the immune system’s disease fighters—new genetic instructions to recognize and destroy cancer cells. These instructions come in the form of chimeric antigen receptors (CARs), molecules that protrude from the surface of T cells. The CAR binds to a target on a cancer cell surface, triggering the T cell to attack and kill the tumor cell. Seven CAR T cell therapies are currently approved by FDA for use in blood cancers. “Engineered T cells, reprogrammed to express chimeric antigen receptors (CAR) or T cell receptors (TCR), have transformed cancer treatment and are being explored as therapeutics for autoimmune and infectious diseases,” the authors commented.
However, accessing these therapies, which cost $400,000 to $500,000, has proven difficult for many patients. The manufacturing process requires specialized facilities and takes weeks. Before receiving the engineered cells, patients must also undergo intensive chemotherapy that some patients can’t tolerate.
“It’s become a global access issue; many patients who would benefit from CAR T cells either can’t afford them or can’t get them fast enough,” Eyquem said. Re-engineering immune cells in the body—in vivo manufacturing—could completely bypass the costly and complex ex vivo manufacturing, while also eliminating the need for preparatory chemotherapy. Eyquem told GEN, “In vivo CAR T cell generation aims to engineer T cells directly within the body … Done without lymphodepleting preconditioning, this approach has the potential to dramatically reduce cost, shorten timelines, and broaden global access to a treatment that remains out of reach for the vast majority of patients who might benefit from it.”
Dual-vector system
For their newly reported research, Eyquem and collaborators, including scientists at the Gladstone Institutes, Duke University, and Innovative Genomics Institute, designed a dual-particle system to carry CRISPR-Cas9 ribonuclear protein (RNP) gene-editing machinery—the molecular scissors required to alter genes—directly to T cells circulating in the body. “Using CRISPR–Cas9 and adeno-associated virus (AAV)-mediated homology-directed repair (HDR), we targeted CAR integration into the endogenous human TCR alpha locus (TRAC),” the authors summarized in their paper.
William Nyberg, PhD, co-first author and former postdoc in the Eyquem lab, further explained, “We developed a two-vector system to deliver CRISPR–Cas9 ribonucleoproteins and a DNA donor template, using enveloped delivery vehicles and adeno-associated viruses, respectively … We optimized both vectors for T cell-specific delivery and gene-targeting efficiency.”
The goal, Eyquem explained, was to remain both cell-specific and locus-specific, using CRISPR to integrate the CAR transgene at a defined site in the T cell genome. “That required delivering two distinct components to the same T cell in vivo,” he told GEN, “… the CRISPR machinery to cut at a precise location, and a DNA template encoding the CAR to be inserted there … Because these two payloads have fundamentally different delivery requirements, we developed a two-vector system, with each vector specialized for one component.”
Eyquem expanded on the process for developing the two vectors. “The first vector—for delivery of the CRISPR–Cas9 machinery—was developed by Jennifer Hamilton in the Doudna laboratory. We call it an Enveloped Delivery Vehicle, or EDV. It packages a fully formed Cas9–gRNA complex inside a particle that outwardly resembles a lentivirus, but instead of delivering DNA it delivers the editing protein directly. By engineering its surface with a mutated fusogen and an anti-CD3 antibody fragment, we direct it selectively to T cells in vivo.
“The second vector carries the CAR DNA template. We used an AAV, a vector naturally suited to efficient nuclear DNA delivery … The challenge was that the standard AAV serotype used in this context, AAV6, is poorly suited for in vivo use … In collaboration with Aravind Asokan at Duke, we evolved a new capsid variant (AAV-hT7) that overcomes barriers … Bernard further demonstrated using a genome wide screen that our engineered AAV interacts with CD7.”
While a two-vector system might sound more complex, it carries a critical advantage, Eyquem explained. “… while neither vector alone is entirely T cell-specific, their combination is. Only cells that take up both the EDV and AAV-hT7 undergo CAR integration and that overlap is highly specific to T cells. Engineering precision is preserved through the intersection of the two vectors working together.”
Nyberg, who now has his own lab at the Karolinska Institute in Sweden, was the first to combine both vectors in a humanized mouse model to deliver a CD19 CAR, Eyquem noted. “Remarkably, after a single injection of the two vectors, we observed a large population of CAR T cells and complete B cell aplasia … The day he analyzed the cells, I told him: ‘Will, let’s have a drink, this is going to be BIG.’ It has been working in every system and model we have tested since.”
In their reported study the researchers tested the technology in mice engrafted with aggressive leukemia. A single injection of the dual-particle system cleared all detectable cancer in nearly all the mice within two weeks. The engineered CAR T cells made up as much as 40% of immune cells in some organs and successfully eliminated cancer from both the bone marrow and spleen.
Potential for solid tumor therapy
The approach also worked against multiple myeloma and, strikingly, against a solid sarcoma tumor. “Our demonstration that in vivo generated CAR T cells can control an aggressive solid tumor model in mice, something many in the field assumed would be beyond reach has given us real confidence that this is worth pursuing seriously,” Eyquem said.
“One of the most important findings was a direct head-to-head comparison with engineered lentiviral vectors, the leading competing approach for in vivo CAR T cell generation,” he continued. “At an equivalent dose, our TRAC-integrated CAR T cells were markedly superior. We believe this comes down to the fundamental difference between site-specific and random integration. With site-specific integration at the TRAC locus, every successfully engineered T cell expresses a functional, physiologically regulated level of CAR. With lentiviral delivery, expression is lower on average and highly heterogeneous, suggesting that only a subset of transduced cells reaches the threshold needed for optimal antitumor function. Precision of integration translates directly into potency.”
Fundamentally different
The researchers’ system is fundamentally different from other integrative in vivo platforms, he stated. “Other integrative in vivo platforms currently being tested clinically deliver fully independent expression cassettes, meaning the CAR gene carries its own promoter and can be expressed in any cell the vector happens to transduce. This creates real risks: if a tumor cell is accidentally transduced, for example, it will express the CAR and can downregulate the target antigen on its surface, driving antigen-negative relapse and treatment failure.
“We deliver what is called a promoterless cassette, a CAR transgene that carries no promoter of its own and therefore cannot be expressed unless it integrates precisely and in-frame into the right gene in the right cell type. That gene is TRAC, which encodes one of the chains of the T cell receptor and is exclusively expressed in T cells. This provides a level of selectivity that no randomly integrating vector can match.”
Beyond CAR T cell therapy
The choice of TRAC as the integration site offers several advantages beyond simple T cell specificity, he said. “We believe this represents a breakthrough that extends well beyond CAR T cell therapy. The same fundamental strategy, pairing a cell-targeted RNP delivery vehicle with an evolved AAV carrying a site-specific DNA template, could in principle be applied to other cell types and other indications. The TRAC locus and T cells are our proof of concept, but the platform logic is broadly applicable wherever precise, stable transgene integration in a defined cell population is therapeutically desirable.”
Eyquem and collaborators have established Azalea Therapeutics to advance the dual-vector platform. Azalea has exclusively licensed foundational academic IP to support its programs for in vivo site-specific engineering.
The reported mouse data have given the researchers the confidence that the platform is “… ready to be pushed toward patients, and that is now the primary driving force behind everything we do,” Eyquem said. Azalea has already moved with “remarkable speed” since its founding, he stressed. “The team onboarded the technology, reproduced all the key findings from the Nature paper, and has since further developed and scaled the platform for clinical application.”
A critical milestone in that journey was demonstrating efficacy in non-human primates, a pivotal translational step that the mouse data alone could not provide,” he explained to GEN. “In a proof-of-concept non-human primate study evaluating feasibility, pharmacodynamics and tolerability, a single intravenous dose of the EDV and AAV vectors achieved complete B cell aplasia in peripheral blood by day 10, expansion of TRAC-CAR T cells to approximately 35% of all peripheral T cells confirming robust in vivo engineering, complete B cell aplasia in lymph nodes and bone marrow by day 13, and a favorable tolerability profile with no unexpected findings to date.”
These data were presented at the ASGCT Breakthroughs in Targeted In vivo Gene Editing session back in November 2025, he said.
Towards the clinic
Azalea is now “racing toward the clinic, with CD19-directed CAR T cell therapy as the lead program,” GEN learned. “… The goal is clear: to get this technology to patients as fast as responsibly possible … The most likely first indication is a CD19-directed CAR T cell therapy, pursued across two distinct disease areas: B cell malignancies and autoimmune disorders.”
The current plan is to reach first-in-man by the end of 2027, Eyquem suggested. “Given where the platform stands today with compelling mouse data, non-human primate studies already demonstrating robust in vivo CAR T cell generation and deep B cell aplasia with a favorable tolerability profile, and Azalea actively working through the necessary manufacturing and regulatory steps, we believe this is an ambitious but realistic timeline.”
The post Dual-Vector System Generates CAR T Cells <i>In Vivo</i> appeared first on GEN – Genetic Engineering and Biotechnology News.
GEN – Genetic Engineering & Biotechnology News
Bioengineered Implants Deliver Multi-Drug Therapy in Animal Models
In a new paper, scientists from Northwestern University and their collaborators at Rice University and Carnegie Mellon University report on their progress towards developing so-called implantable “living pharmacies.” These are tiny devices containing engineered cells that continuously produce medicines inside the body. Details of the study, which was done in rats, are published in Device in a paper titled “Design of a wireless, fully implantable platform for in-situ oxygenation of encapsulated cell therapies.”
The device, which is called the hybrid oxygenation bioelectronics system for implanted therapy or HOBIT, is roughly the size of a folded stick of gum. It integrates engineered cells with oxygen-producing bioelectronics and is designed in such a way that the cells are shielded from the body’s immune system while also receiving oxygen and nutrients needed to keep them alive and producing drugs for several weeks. In the future, these devices could be deployed to treat chronic conditions without requiring patients to carry, inject, or remember to take medications.
“This work highlights the broad potential of a fully integrated biohybrid platform for treating disease,” said Jonathan Rivnay, PhD, a professor of biomedical engineering and materials science and engineering at Northwestern and a co-principal investigator of the project. “Traditional biologic drugs often have very different half-lives, so maintaining stable levels of multiple therapies can be challenging. Because our implanted ‘cell factories’ continuously produce these biologics, keeping the cells alive with our oxygenation technology allows us to sustain steady levels [of] multiple different therapeutics at once.”
Solving the oxygenation challenge was critical to the success of HOBIT. When engineered cells are packed together in an implant, they compete for oxygen to live. Without a steady supply, many cells die, which limits how much medicine the implants can produce. In an earlier study, Rivnay and his collaborators demonstrated how a tiny electrochemical device could generate oxygen by splitting nearby water molecules, and showed that supplying oxygen locally dramatically improved the survival of implanted therapeutic cells. The latest iteration of their device integrates that oxygen-generation technology in a fully implantable, wireless system.
Digging into the details of the device, HOBIT contains three primary components: a cell chamber that holds the genetically engineered cells, a miniature oxygen generator, and electronics and a battery to regulate oxygen production and wirelessly communicate with external devices. Because the device produces oxygen directly inside the implant, the cells receive a steady supply even in hypoxic environments. “We are producing oxygen directly where the cells need it,” Rivnay said. “That allows us to support much higher cell densities in a much smaller space.” In fact, “cell densities in HOBIT were roughly six times higher than conventional unoxygenated encapsulation approaches.”
According to the paper, the team engineered the cells to produce three different biologics—an anti-HIV antibody, a GLP-1-like peptide used to treat type 2 diabetes, and leptin, a hormone that regulates appetite and metabolism. They implanted the devices under the skin of rats and monitored drug levels in their bloodstreams for 30 days. Blood measurements of animals with the implanted devices showed sustained levels of all three biologics throughout the study period. In contrast, in animals that were implanted with devices without oxygenation, the biologics that had shorter half-lives were undetectable by the seventh day. Drugs with longer half-lives in these animals also declined steadily over time. At the end of the testing period, roughly 65% of the cells in the oxygenated devices remained viable compared with roughly 20% in control devices.
For their next steps, the scientists intend to test their devices in larger animal models and explore disease-specific applications, including therapies based on transplanted pancreatic cells. “As these technologies continue to develop, devices like this could eventually act as programmable drug factories inside the body—delivering complex therapies in ways that simply aren’t possible today,” Rivnay said.
The post Bioengineered Implants Deliver Multi-Drug Therapy in Animal Models appeared first on GEN – Genetic Engineering and Biotechnology News.
GEN – Genetic Engineering & Biotechnology News
Gut-Immune Link Identified in Multiple Sclerosis-Related Neuroinflammation
Multiple sclerosis (MS) is a debilitating neurological disorder caused by malfunctioning immune responses that target the brain and spinal cord of the central nervous system (CNS). New research led by Shohei Suzuki, MD, PhD, assistant professor, division of gastroenterology and hepatology, and Tomohisa Sujino, PhD, associate professor, School of Medicine, at Keio University, Japan, has now indicated how the gut can initiate neuroinflammation in multiple sclerosis.
Their study found that intestinal epithelial cells (IECs) promote the development of pathogenic T cells that migrated to the spinal cord and induced disease symptoms in mouse models of the disorder.
The researchers examined intestinal tissues from patients with MS and mice with experimental autoimmune encephalomyelitis (EAE), a close analog of MS. In both cases, they observed an increase in TH17 cells and an upregulation of major histocompatibility complex class II (MHC II) expression in IECs. Deleting MHC II in IECs reduced the accumulation of TH17 cells in the gut and lowered the severity of EAE. They suggest the results could inform future strategies for developing targeted therapeutics against autoimmunity.
“While current therapies for MS often target B cells, our study highlights the gut as an important therapeutic site,” Suzuki commented. “Modulating intestinal microbiota or antigen-presenting activity of IECs represents new approaches to treating autoimmune neurological diseases.”
Suzuki, Sujino, and colleagues reported on their findings in Science Immunology, in a paper titled “Intestinal Epithelial MHC Class II Induces Encephalitogenic CD4⁺ T Cells and Initiates Central Nerves System Autoimmunity,” in which they concluded, “Our findings reveal an interaction between gut IECs and neuroinflammatory diseases through MHC II expression in human MS and mouse EAE, providing a mechanistic link between gut immune education and CNS autoimmunity and opening new avenues for targeting intestinal immunity in neuroinflammatory diseases.
Failure of the immune system to distinguish ‘self’ from ‘non-self’ entities leads to excessive autoimmune responses against self-proteins like myelin, which forms a protective covering on the neurons. Multiple factors influence the onset and progression of MS, including genetic susceptibility, environmental triggers, and, more recently, the gut microenvironment. Patients with MS exhibit alterations in their gut microbiota, while the gut microbiota and microbial metabolites play a pivotal role in shaping the chronic autoreactive immune responses. “… in an experimental autoimmune encephalomyelitis (EAE) model, commensal or specific microbes were found to be essential for disease initiation and progression,” the authors wrote.
However, in trying to define this gut–CNS axis, the cellular mechanisms that relay the gut-derived signals to the immune system to influence autoimmune inflammation in the CNS remain poorly understood. “Increasing evidence shows that the gut microbiota influences neurological diseases such as Parkinson’s, Alzheimer’s, and MS,” Sujino stated. “However, the mechanisms linking gut microbes, intestinal immunity, and brain inflammation remain unclear. We were keen to identify how gut immune responses contribute to neuroinflammatory diseases.”
Prior research has shown that gut-derived signals can promote the differentiation of T cells into pathogenic T helper 17 (TH17) in mouse models of MS. Recent studies have suggested that IECs can function as antigen presenting cells that help induce these pathogenic cells, but the underlying mechanisms have been unclear.
Building on their previous observation that mild intestinal (ileal) inflammation exists in experimental autoimmune encephalomyelitis (EAE), which is a mouse model of MS, the authors set out to test whether similar inflammation is present in patients with MS. By performing single-cell RNA sequencing on intestinal biopsies, the team identified that inflammatory Th17 cells accumulate in the mouse EAE model as well as in the intestine of patients with MS, suggesting a conserved gut–CNS axis that may be active in human diseases.
In both EAE mice and patients with MS, intestinal epithelial cells upregulated antigen presentation pathways. Particularly, epithelial cells in the ileum had higher expression of major histocompatibility complex class II (MHC II) that presents antigens to CD4+ T cells. “Clinically, patients with MS exhibited an increased expression of epithelial MHC II–associated genes and an accumulation of CD4 T cells in the small intestine, suggesting the conservation of this gut-CNS axis in human diseases,” the scientists stated. Experiments showed that selective deletion of MHC II in IECs reduced pathogenic Th17 cell generation and disease severity. “Conditional deletion of MHC II in IECs showed that epithelial antigen presentation was indispensable for the local expansion of pathogenic Th17 cells in the gut and their subsequent migration to the CNS,” the team stated.
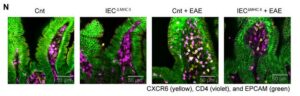
![Immunofluorescence analysis was performed on terminal ileum samples from Cnt, IECΔMHCII, Cnt + EAE, and IECΔMHCII + EAE mice. A total of 3–5 tissue sections were analyzed per mouse, with 3 mice included in each group. [Shohei Suzuki]](https://www.genengnews.com/wp-content/uploads/2026/03/low-res-2-1-300x96.jpeg)
mouse, with three mice included in each group. [Shohei Suzuki]
IECs do not typically present antigens to immune cells. So, the team conducted co-culture assays to test the antigen presentation function of IECs. Their findings demonstrate that IECs can directly present antigens in an MHC II-dependent manner to prime CD4+ T cells in the gut. Notably, in these assays, IECs induced Th17 polarization of activated CD4+ T cells. It became clear that the gut was a critical site for immune activation of pathogenic CD4+ T cells that polarized into pro-inflammatory Th17 cells. “These findings provide direct functional evidence that IEC-expressed MHC II is sufficient to drive Th17 polarization from primed CD4 T cells in an antigen-dependent manner, supporting a direct role for IECs as non-professional antigen-presenting cells,” the scientists reported.
To investigate whether the Th17 cells directly contribute to the pool of autoreactive cells in the CNS, they used transgenic mice that express the Kaede protein, which undergoes photoconversion from green to red fluorescence upon exposure to violet light. This model allowed for precise tracking of pathogenic Th17 cells induced in the intestinal lamina propria that then migrate to the spinal cord and drive neuroinflammation.
Taken together, the study findings reveal a critical role for MHC II expressed by IECs in the expansion of pathogenic Th17 cells that subsequently migrate to the CNS during EAE, providing a mechanistic link between gut immune responses and autoimmune neuroinflammatory diseases. The results demonstrate that while systemic circulation allows T cell exchange across immune tissues, the epithelial–immune interactions within the gut mucosal compartment can essentially shape effector T cell responses in the brain.
“This study reveals a previously unknown role of IECs in antigen presentation and Th17 programming, thereby defining a gut-CNS immunological axis with important implications for understanding and treating autoimmune neuroinflammation,” the authors concluded. “Our findings suggest that the modulation of epithelial antigen presentation could serve as a novel therapeutic approach for MS and related diseases. Given the accessibility of the gut epithelium to dietary, microbial, and pharmacological interventions, targeting IEC–T cell interactions may offer a tractable strategy for immunomodulation.”
The post Gut-Immune Link Identified in Multiple Sclerosis-Related Neuroinflammation appeared first on GEN – Genetic Engineering and Biotechnology News.
GEN – Genetic Engineering & Biotechnology News
Agentic AI, Virtual Cell, LNP Vaccine Boosters, Engineered Organs, and Mergers
This week, agentic AI steps into the limelight buoyed by the momentum from generative AI. And there’s a new virtual cell model in town courtesy of AI-drug developer Xaira Therapeutics. From the frontiers of AI, our discussion turned to feats of engineering in regenerative medicine and lipid nanoparticles. In one study, scientists redesigned LNPs to avoid the liver and accumulate in the lymph nodes. In the other, efforts to develop and implant a lab grown esophagus from donor pigs bear fruit. Finally, Novartis plans to spend up to $3 billion to expand its cancer pipeline with the acquisition of Pikavation Therapeutics. And Merck is acquiring Terns Pharmaceuticals for approximately $6.7 billion also with an eye towards boosting its cancer portfolio.
Listed below are links to the GEN stories referenced in this episode of Touching Base:
NVIDIA GTC 2026: Agentic AI Inflection Hits Healthcare and Life Sciences
By Fay Lin, PhD, GEN Edge, March 18, 2026
Xaira’s First Virtual Cell Model Is Largest To-Date, Toward Complex Biology
By Fay Lin, PhD, GEN Edge, March 25, 2026
Modified Lipid Nanoparticles Boost mRNA Vaccine Delivery to Lymph Nodes
GEN, March 24, 2026
Engineered Esophagus Rebuilds Missing Organ Segment in Pig Models
GEN, March 20, 2026
Novartis Acquires Pikavation for Up to $3B, Expanding Cancer Pipeline
GEN, March 22, 2026
Merck Bolsters Cancer Pipeline with $6.7B Terns Buyout
By Alex Philippidis, GEN Edge, March 25, 2026
Touching Base Podcast
Hosted by Corinna Singleman, PhD
Behind the Breakthroughs
Hosted by Jonathan D. Grinstein, PhD
The post Agentic AI, Virtual Cell, LNP Vaccine Boosters, Engineered Organs, and Mergers appeared first on GEN – Genetic Engineering and Biotechnology News.


Illinois’ financial crisis could bring the state to a halt
The final 6 ‘Game of Thrones’ episodes might feel like a full season
New Season 8 Walking Dead trailer flashes forward in time
Mod turns ‘Counter-Strike’ into a ‘Tekken’ clone with fighting chickens
Meet Superman’s grandfather in new trailer for Krypton
Disney’s live-action Aladdin finally finds its stars

STAT+: FDA approves Sanofi diabetes drug for children with stage 3 diabetes
Opinion: ‘I’m pretty much all in’: An interview with a woman starting medical residency at almost 73

STAT+: Trump administration revisits policy to close Medicare drug price negotiation loophole
Lilly unveils first clinical data behind $2.3B Ajax deal, showing JAK inhibitor works ‘right out of the gate’
Hantavirus One-Shot mRNA Vaccine Fully Protects in Syrian Hamster Model
SonoThera Raises $125M to Develop Ultrasound-Mediated Genetic Medicines
Illinois’ financial crisis could bring the state to a halt
The final 6 ‘Game of Thrones’ episodes might feel like a full season
New Season 8 Walking Dead trailer flashes forward in time
Mod turns ‘Counter-Strike’ into a ‘Tekken’ clone with fighting chickens
Meet Superman’s grandfather in new trailer for Krypton
Disney’s live-action Aladdin finally finds its stars
-
 Uncategorized9 years ago
Uncategorized9 years agoThese ’90s fashion trends are making a comeback in 2017
-
Uncategorized9 years ago
According to Dior Couture, this taboo fashion accessory is back
-
Endpoints News3 months ago
Novartis to pay $2B upfront to take next-gen PI3Kα inhibitor from Synnovation
-
Uncategorized9 years ago
Phillies’ Aaron Altherr makes mind-boggling barehanded play
-
Uncategorized9 years ago
Uber and Lyft are finally available in all of New York State
-
Contributors9 years ago
The final 6 ‘Game of Thrones’ episodes might feel like a full season
-
Uncategorized9 years ago
Steph Curry finally got the contract he deserves from the Warriors
-
Uncategorized9 years ago
The old and New Edition cast comes together to perform